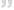FGFR异常激活与多种肿瘤相关
成纤维细胞生长因子受体2(FGFR2)是一种受体酪氨酸激酶(RTK),属于FGFR家族,该家族包括四种高度保守的受体FGFR1-FGFR4,这些受体均具有单通道跨膜蛋白结构,包含胞外、跨膜和胞内酪氨酸激酶结构域。FGF/FGFR信号通路主要涉及成纤维细胞生长因子(FGF)与受体FGFR的结合、受体二聚化以及随后的细胞内激酶自动磷酸化级联反应,该通路在细胞增殖、分化、凋亡和迁移方面发挥着关键作用,若异常激活可诱发多种疾病。
异常的FGFR基因改变被认为是多种实体瘤肿瘤细胞增殖的驱动因素,约1.9%-7.1%的肿瘤患者中存在FGFR异常。按肿瘤类型来看,FGFR异常改变在尿路上皮癌、结直肠癌、胃癌、乳腺癌等肿瘤中的发生频率较高。按基因组改变类型来看,FGFR异常大多是基因扩增(53.7%-66%),其次是突变(26%-38.8%)和重排/融合(5.6%-8%)。按受体种类来看,FGFR1、FGFR2、FGFR3和FGFR4的畸变频率分别为49%-56.8%、14.2%-19%、17.7%-26%以及2.8%-7%。
FGFR抑制剂开发:克服耐药性、提升安全性
目前,有多种针对FGFR通路的疗法正在开发中,根据其作用机制可以分为小分子TKI、单抗、ligand trap以及ADC。
小分子抑制剂是研发进展最快的药物类型,目前已有五种选择性FGFR小分子抑制剂获批用于治疗FGFR突变的肿瘤患者。其中,非共价可逆选择性抑制剂(如已上市的erdafitinib、infigratinib和pemigatinib)可作用于Hinge region附近的药物结合口袋,易受Gatekeeper(常位于ATP结合口袋的入口处)、Molecularbrake(常与激酶的活性调节有关)或DFG-latch(其构象变化可以影响ATP结合口袋的开关)突变的影响;而共价不可逆抑制剂(futibatinib、lirafugratinib等),与位于P loop的保守半胱氨酸残基(FGFR2中为C491)相互作用,受这些改变影响的可能性相对较小。
FGFR1脱靶抑制导致的高磷血症、FGFR4介导的腹泻等副作用仍然限制了部分药物的临床获益。因此,选择性靶向FGFR2/3及其耐药突变可能克服相关突变介导的耐药性并减少药物的副作用。



 查看资料
查看资料 收藏
收藏 顶
顶  踩
踩